一种基于外显子或基因组测序数据诊断脊髓性肌萎缩(SMA)的工具

项目描述
SMA Finder
SMA Finder是一个基于Illumina外显子、基因组或靶向测序数据诊断脊髓性肌萎缩(SMA)的工具。
它以参考序列(FASTA)和1个或多个比对文件(CRAM或BAM)作为输入,评估SMN1和SMN2在c.840位点的读取,以检测SMA最常见的分子原因,并报告是否发现了SMN1的完全丢失。
它已在短读数据上进行了测试和确认,这些数据与GRCh37、GRCh38或T2T对齐,并使用BWA对齐器进行对齐,准确性非常高。
限制
- 不报告SMA携带者状态或SMN1/SMN2拷贝数
- 不检测由不涉及c.840位点的SMN1失活突变引起的约5%的病例
- 为了做出调用,至少需要14个读取重叠SMN1和SMN2中的c.840位置
- 是在来自全血DNA的Illumina短读测序数据上开发和测试的。在其他测序技术、样本类型和对齐管道中的性能未知。
安装
python3 -m pip install sma-finder
示例
示例命令
sma_finder --verbose --hg38-reference-fasta /ref/hg38.fa sample1.cram
命令输出
Input args:
--hg38-reference-fasta: /ref/hg38.fa
--output-tsv: sample1.sma_finder_results.tsv
CRAMS or BAMS: sample1.cram
---
Output row #1:
filename_prefix sample1
file_type cram
genome_version hg38
sample_id s1
sma_status has SMA
confidence_score 168
c840_reads_with_smn1_base_C 0
c840_total_reads 174
Wrote 1 rows to sample1.sma_finder_results.tsv
用法
用法帮助文本
sma_finder --help
usage: sma_finder.py [-h] [--hg37-reference-fasta HG37_REFERENCE_FASTA]
[--hg38-reference-fasta HG38_REFERENCE_FASTA]
[--t2t-reference-fasta T2T_REFERENCE_FASTA]
[-o OUTPUT_TSV] [-v]
cram_or_bam_path [cram_or_bam_path ...]
positional arguments:
cram_or_bam_path One or more CRAM or BAM file paths
optional arguments:
-h, --help show this help message and exit
--hg37-reference-fasta HG37_REFERENCE_FASTA
HG37 reference genome FASTA path. This should be
specified if the input bam or cram is aligned to HG37.
--hg38-reference-fasta HG38_REFERENCE_FASTA
HG38 reference genome FASTA path. This should be
specified if the input bam or cram is aligned to HG38.
--t2t-reference-fasta T2T_REFERENCE_FASTA
T2T reference genome FASTA path. This should be
specified if the input bam or cram is aligned to the
CHM13 telomere-to-telomere benchmark.
-o OUTPUT_TSV, --output-tsv OUTPUT_TSV
Optional output tsv file path
-v, --verbose Whether to print extra details during the run
输出
输出文件 .tsv 每行对应一个输入的 CRAM 或 BAM 文件,包含以下列:
| filename_prefix | CRAM 或 BAM 文件名前缀。如果输入文件是 /path/sample1.cram,则此前缀为 "sample1"。 |
| file_type | "cram" 或 "bam" |
| genome_version | "hg37","hg38" 或 "t2t" |
| sample_id | 来自 CRAM 或 BAM 文件头的 sample id(从读组元数据中解析得出) |
| sma_status | 可能的值为 "has SMA" "does not have SMA" "SMN c.840 位置覆盖不足" |
| confidence_score | PHRED 比例的整数分数,衡量 sma_status 正确的置信水平。分数越高,置信度越高。其计算方式与 GATK HaplotypeCaller 基因型中的 PL 字段类似。 |
| c840_reads_with_smn1_base_C | 在 SMN1 加 SMN2 的 c.840 位置具有 'C' 核苷酸的读取数量 |
| c840_total_reads | 重叠在 SMN1 加 SMN2 的 c.840 位置的读取总数 |
合并多个样本的结果
在许多样本上运行 SMA Finder 后,通常需要将每个样本的输出表合并为一个单一的表格。以下 shell 命令是一种合并方式:
combined_table_filename=combined_results.tsv
head -n 1 $(ls *.tsv | head -n 1) > ${combined_table_filename} # get table header from the 1st table
for i in *.tsv; do
tail -n +2 $i >> ${combined_table_filename} # concatenate all tables
done
绘制合并结果
可以使用 plot_SMN1_SMN2_scatter 命令生成总结多个样本读取计数的散点图
python3 plot_SMN1_SMN2_scatter.py --format svg --format png ${combined_table_filename}
它生成类似于以下图表的图形,该图表基于一个包含 16,626 个外显子的神经肌肉队列
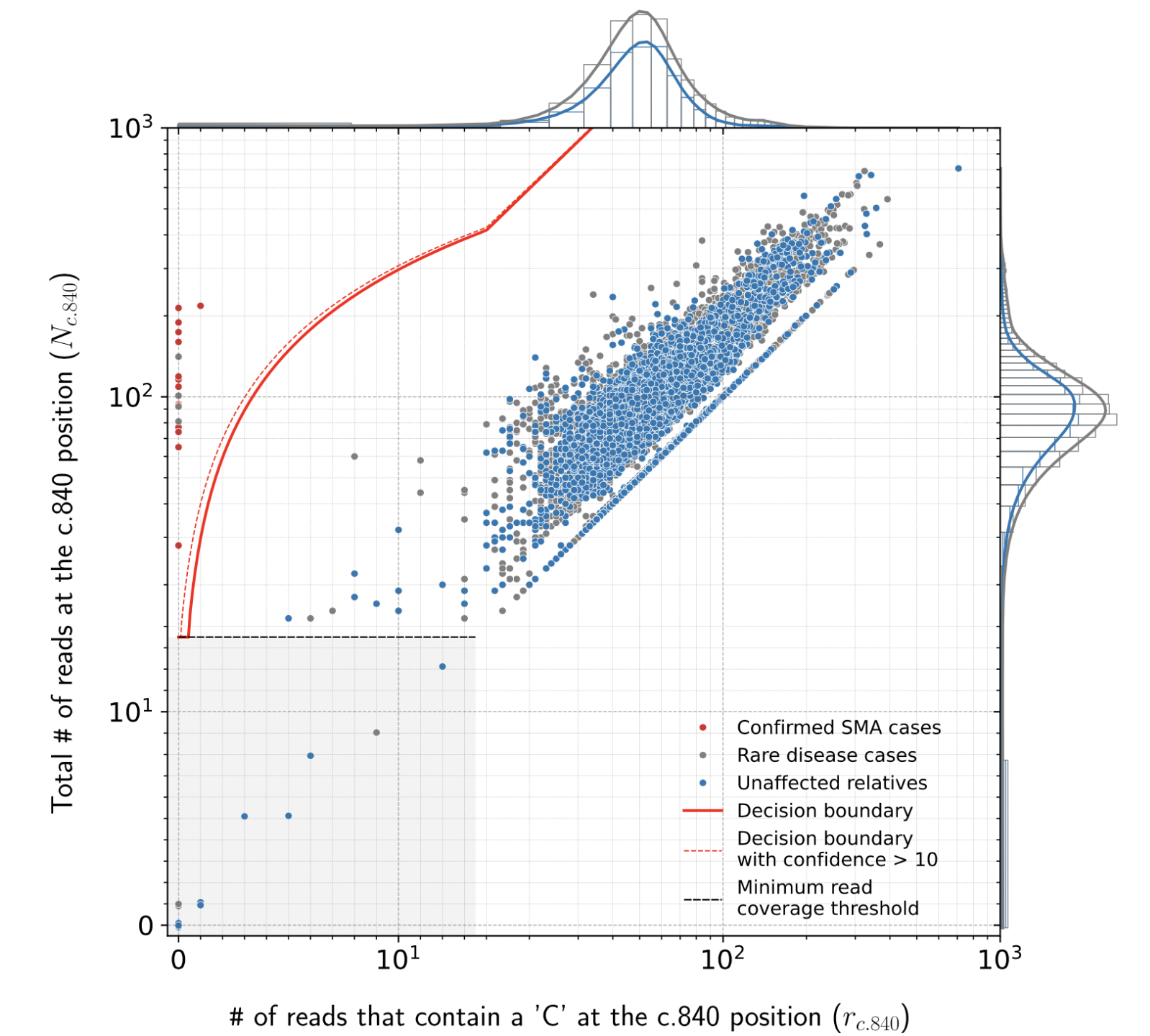
详细信息
本 SMA Finder 海报在 SVAR22 会议中展出


下载文件
下载适用于您平台的文件。如果您不确定选择哪个,请了解更多关于 安装包 的信息。
源分布
sma_finder-1.4.4.tar.gz (13.6 kB 查看哈希值)
构建分布
sma_finder-1.4.4-py3-none-any.whl (14.2 kB 查看哈希值)
关闭
sma_finder-1.4.4.tar.gz 的哈希值
| 算法 | 哈希摘要 | |
|---|---|---|
| SHA256 | e0fdad3e594d1d909fc1f1be91a17464bb64dd0f86096da4b2bb66c4fc92f963 |
|
| MD5 | c13d70376982e9f431ebd701655f8b69 |
|
| BLAKE2b-256 | 63594b9cb498a264ca79ffe6ac60a3ef21fc83976e6782c5ae184e7341c8ebb7 |
关闭
sma_finder-1.4.4-py3-none-any.whl 的哈希值
| 算法 | 哈希摘要 | |
|---|---|---|
| SHA256 | f3f7eb50a71921560194e16c1a8434078a6b9eef5bff8456bb13475b6e05e18f |
|
| MD5 | 18142d8947d47122328246e7776f1413 |
|
| BLAKE2b-256 | 630f46dfd349199673b74b45708c838729b7db3a6dee53fcdad8d7b219527dfd |







