组成扰动自编码器 (CPA)


项目描述
CPA - 组成扰动自编码器



什么是CPA?
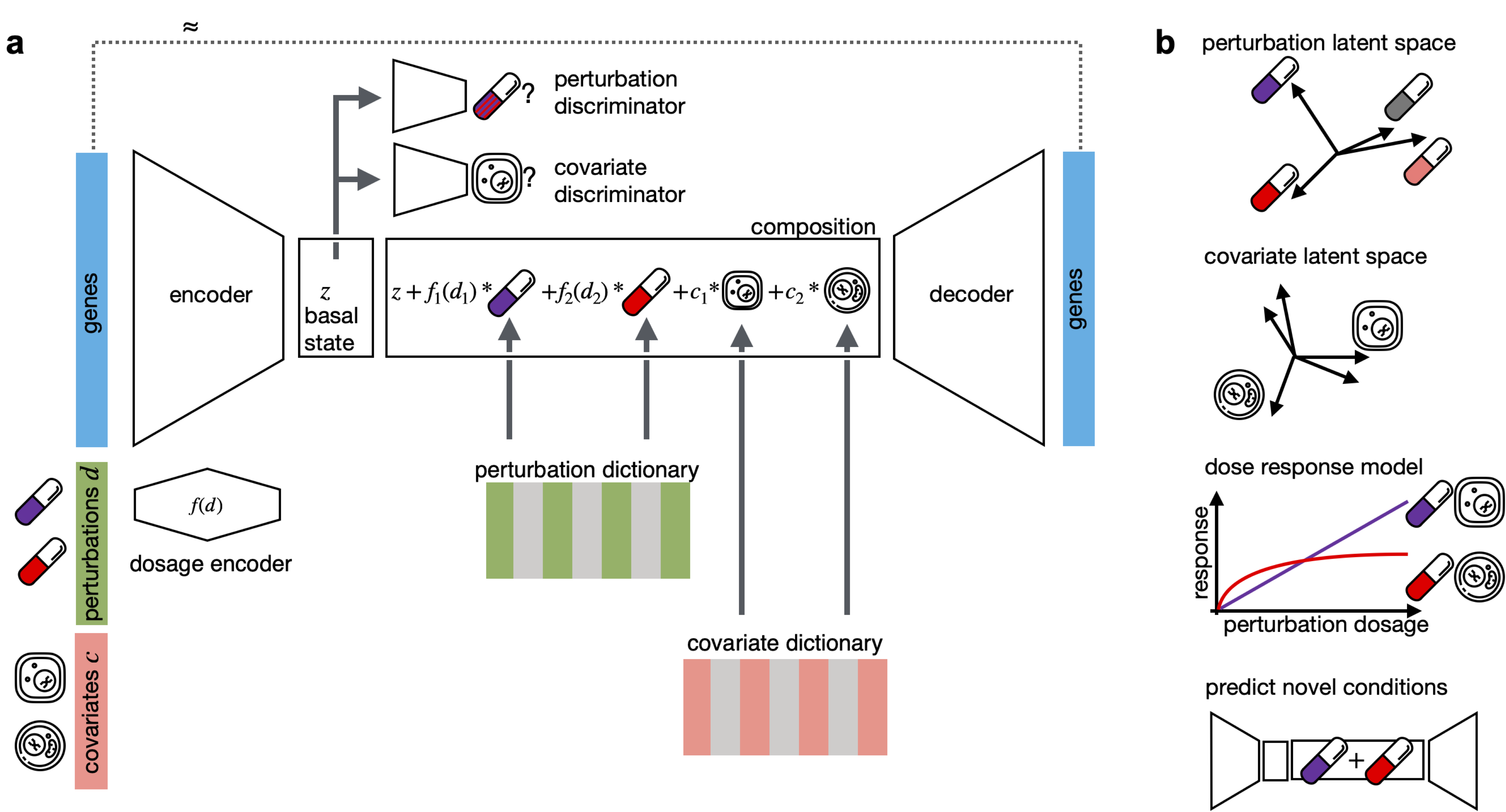
CPA是一个学习单细胞层面扰动影响的框架。CPA编码并学习不同细胞类型、剂量和组合的表型药物反应。CPA允许
- 预测未见过的药物和基因组合在不同剂量和不同细胞类型中的分布外的预测。
- 学习可解释的药物和细胞类型潜在空间。
- 估计每个扰动及其组合的剂量-反应曲线。
- 将扰动效应从一种细胞类型转移到未见的细胞类型。
- 在潜在空间和基因表达空间上实现批量效应去除。
安装
安装CPA
您可以使用pip安装CPA,也可以直接从github安装以访问最新开发版本。请参阅详细说明此处。
如何使用CPA
有一些教程此处可以帮助您开始使用CPA。以下表格包含教程列表
| 描述 | 链接 |
|---|---|
| 预测组合药物扰动 |  - -  |
| 使用外部嵌入预测未见过的扰动,使模型能够预测未见过的药物的反应 | - |
| 预测组合CRISPR扰动 | - |
| 上下文迁移演示(例如,预测扰动(如疾病)对未见过的细胞类型的影响,或将扰动效应从一个上下文转移到另一个上下文)——基于IFN-β单细胞RNA测序扰动数据集 | - |
| 基因表达和潜在空间中的批次效应去除 | - |
如何优化您数据中的CPA超参数
我们提供了一个示例脚本,用于使用CPA(基于scvi-tools超参数优化器)内置的超参数优化功能。您可以在examples/tune_script.py中找到脚本。
使用tune_script.py完成超参数优化后,result_grid.pkl将使用pickle库保存到您的当前目录。您可以使用以下代码加载结果
import pickle
with open('result_grid.pkl', 'rb') as f:
result_grid = pickle.load(f)
从这里,您可以按照Ray文档中的说明分析运行,并选择最适合您数据的最优超参数。
您还可以使用与wandb的集成来记录超参数优化结果。您可以在examples/tune_script_wandb.py中找到脚本。--> use_wandb=True
所有这些都基于Ray Tune。您可以在Ray Tune文档中找到有关超参数优化的更多信息。
该调整器是从scvi-tools v1.2.0(未发布)发布说明中适配和调整的
数据集和预训练模型
数据集和预训练模型可在此处获取。
预处理自定义scRNAseq扰动数据集的食谱
如果您可以访问您的原始数据,您可以按照以下步骤预处理您的数据集。原始数据集应是一个包含原始计数和所需元数据(例如,扰动、剂量等)的scanpy对象。
预处理步骤
-
检查细胞元数据中的所需信息:a) 扰动信息应在
adata.obs中。b) 剂量信息应在adata.obs中。在CRISPR基因敲除、疾病状态、时间扰动等情况下,您可以在adata.obs中创建并添加一个虚拟剂量。例如adata.obs['dosage'] = adata.obs['perturbation'].astype(str).apply(lambda x: '+'.join(['1.0' for _ in x.split('+')])).values
c) [如果可用] 细胞类型信息应在
adata.obs中。d) [多批次集成] 批次信息应在adata.obs中。 -
过滤掉计数少的细胞(
sc.pp.filter_cells)。例如sc.pp.filter_cells(adata, min_counts=100)
[可选]
sc.pp.filter_genes(adata, min_counts=5)
-
将原始计数保存到
adata.layers['counts']。adata.layers['counts'] = adata.X.copy()
-
对计数进行归一化(
sc.pp.normalize_total)。sc.pp.normalize_total(adata, target_sum=1e4, exclude_highly_expressed=True)
-
对归一化计数进行对数转换(
sc.pp.log1p)。sc.pp.log1p(adata)
-
高度可变基因选择:有两种选择:1. 使用
sc.pp.highly_variable_genes函数选择高度可变基因。python sc.pp.highly_variable_genes(adata, n_top_genes=5000, subset=True)2. (特别推荐,特别是针对多批次集成场景)使用scIB的高度可变基因选择函数选择高度可变基因。此函数对批次效应更具鲁棒性,可以用于选择多个数据集中的高度可变基因。python import scIB adata_hvg = scIB.pp.hvg_batch(adata, batch_key='batch', n_top_genes=5000, copy=True)
恭喜!现在您的数据集已经准备好与CPA一起使用了。别忘了使用adata.write_h5ad函数保存您的预处理数据集。
支持和贡献
如果您有任何问题或新的架构或模型可以集成到我们的流程中,您可以提交一个问题
参考
如果您的研究中使用了CPA,请考虑引用Lotfollahi等. 2023
@article{lotfollahi2023predicting,
title={Predicting cellular responses to complex perturbations in high-throughput screens},
author={Lotfollahi, Mohammad and Klimovskaia Susmelj, Anna and De Donno, Carlo and Hetzel, Leon and Ji, Yuge and Ibarra, Ignacio L and Srivatsan, Sanjay R and Naghipourfar, Mohsen and Daza, Riza M and
Martin, Beth and others},
journal={Molecular Systems Biology},
pages={e11517},
year={2023}
}


下载文件
下载适合您平台的应用程序文件。如果您不确定选择哪个,请了解有关安装包的更多信息。