CheckV:评估宏基因组组装病毒基因组的质量


项目描述




CheckV是一个完全自动化的命令行管道,用于评估单链病毒基因组的质量,包括识别整合前病毒的宿主污染、估计基因组片段的完整性以及识别闭合基因组。
快速链接
快速入门
安装
CheckV数据库
运行CheckV
工作原理
主要输出文件
已知问题
常见问题
支持脚本
引用
快速入门
运行以下命令以安装CheckV、下载数据库并运行程序。将</path/to/database>和<input_file.fna>替换为正确的文件路径
conda install -c conda-forge -c bioconda checkv
checkv download_database ./
export CHECKVDB=</path/to/database>
checkv end_to_end <input_file.fna> output_directory -t 16
安装
安装CheckV有三种方法
-
使用conda或mamba(推荐):
conda install -c conda-forge -c bioconda checkv=1.0.1 -
使用pip:
pip install checkv -
使用docker:见下文部分
如果您决定通过pip安装CheckV,请确保已安装以下外部依赖项
- DIAMOND (v2.1.8): https://github.com/bbuchfink/diamond
- HMMER (v3.3): http://hmmer.org/
- Prodigal-gv (v2.6.3): https://github.com/apcamargo/prodigal-gv
上述列出的版本是经过适当测试的。已知DIAMOND v2.1.9存在一个问题,应避免使用。
如果您决定通过docker安装CheckV,请注意,docker镜像可能不代表最新版本。以下为拉取和运行镜像的命令
docker pull antoniopcamargo/checkv
docker run -ti --rm -v "$(pwd):/app" antoniopcamargo/checkv end_to_end input_file.fna output_directory -t 16
CheckV数据库
如果您使用conda或pip安装,则需要下载数据库
checkv download_database ./
您需要更新环境或使用-d标志指定CHECKVDB位置
export CHECKVDB=/path/to/checkv-db
一些用户可能希望使用自己的完整基因组更新数据库
checkv update_database /path/to/checkv-db /path/to/updated-checkv-db genomes.fna
一些用户可能希望下载特定版本的数据库。请参阅https://portal.nersc.gov/CheckV/中所有以前的数据库版本的存档。如果选择此途径,则需要手动构建DIAMOND数据库
wget https://portal.nersc.gov/CheckV/checkv-db-archived-version.tar.gz
tar -zxvf checkv-db-archived-version.tar.gz
cd /path/to/checkv-db/genome_db
diamond makedb --in checkv_reps.faa --db checkv_reps
数据库经常更新。您可以在此处跟踪这些更新
https://portal.nersc.gov/CheckV/README.txt
运行CheckV
运行CheckV有两种方式
使用单个命令运行完整流程(推荐)
checkv end_to_end input_file.fna output_directory -t 16
使用单独的命令运行流程中的每个步骤
checkv contamination input_file.fna output_directory -t 16
checkv completeness input_file.fna output_directory -t 16
checkv complete_genomes input_file.fna output_directory
checkv quality_summary input_file.fna output_directory
要查看CheckV程序和选项的完整列表,请使用:checkv -h 和 checkv <program> -h
已知问题
- DIAMOND v2.1.9存在一个问题,会导致核心转储
- 对于0.9.0版本,您可能会遇到某些prodigal任务失败的错误。
- 对于>=0.9.0,如果您遇到错误指出diamond数据库未找到,请使用
checkv download_database重新下载数据库 - 对于v0.8.1,有时conda安装了DIAMOND的较旧版本,导致错误。请确保conda已安装DIAMOND版本>=2.0.9
工作原理
流程可以分解为4个主要步骤
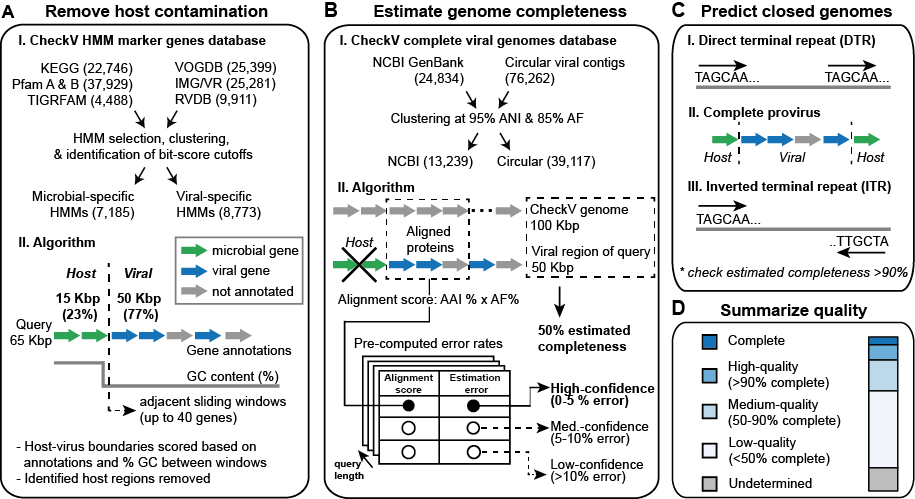
A:从前病毒中去除宿主污染
- 首先根据与自定义HMM数据库的比较,将基因注释为病毒或微生物
- CheckV在contig上扫描(5'到3'),比较相邻基因窗口之间的基因注释和GC含量
- 使用这些信息在每个内基因位置计算分数,并识别宿主-病毒断点
- 最适合大多数为病毒的contig
B:估计基因组完整性
- 首先使用AAI(平均氨基酸同源性)将蛋白质与CheckV基因组数据库进行比较
- 在确定顶级匹配项后,完整性计算为contig长度(或前病毒的病毒区域长度)与匹配参考长度的比率
- 根据对齐强度报告置信水平
- 通常,高和中置信度估计相当准确
- 较少的情况下,您的病毒基因组可能没有与CheckV数据库有密切匹配;在这些情况下,CheckV根据contig上识别的病毒HMM估计完整性
- 根据发现的HMM,CheckV返回基因组完整性的估计范围(例如,35%至60%的完整性),这代表基于具有相同病毒HMM的参考基因组长度分布的90%置信区间
C:预测闭合基因组
-
直接终端重复(DTRs)
- contig起始/结束端的20-bp以上重复序列
- 我们经验中最受信任的签名
- 可能表示环状基因组或从环状模板复制而来的线性基因组(即concatamer)
-
前病毒
- 具有预测的宿主边界在5'和3'端的病毒区域(参见面板A)
- 注意:如果宿主区域已经被移除(例如,使用VIBRANT或VirSorter),CheckV将不会检测到前病毒
-
反转终端重复(ITRs)
- 连续序列 >20-bp 位于 contig 的起始/结束位置(3' 重复是反转的)
- 最不可信的签名
-
对于上述所有方法,CheckV 还会检查 contig 是否根据估计的完整性大约是正确的序列长度;这很重要,因为末端重复可以代表宏基因组组装的伪影
D: 总结质量。
基于 A-C 的结果,CheckV 生成一个报告文件并将查询 contig 分配到五个质量级别之一(与 MIUViG 质量级别一致并在此基础上扩展)
- 完整(见面板 C)
- 高质量(>90% 完整性)
- 中等质量(50-90% 完整性)
- 低质量(<50% 完整性)
- 不确定质量
输出文件
quality_summary.tsv
这包含三个主要模块的综合结果,应该是主要参考的输出。以下是一个示例,以展示您数据中可以预期的结果类型
| contig_id | contig_length | provirus | proviral_length | 基因计数 | 病毒基因 | 宿主基因 | checkv_quality | miuvig_quality | 完整性 | 完整性方法 | 完整基因组类型 | 污染 | kmer_freq | warnings |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 5325 | No | NA | 11 | 0 | 2 | 未确定 | 基因组片段 | NA | NA | NA | 0 | 1 | 未检测到病毒基因 |
| 2 | 41803 | No | NA | 72 | 27 | 1 | 低质量 | 基因组片段 | 21.99 | 基于 AAI(中等置信度) | NA | 0 | 1 | 标记 DTR |
| 3 | 38254 | Yes | 36072 | 54 | 23 | 2 | 中等质量 | 基因组片段 | 80.3 | 基于 HMM(下限) | NA | 5.7 | 1 | |
| 4 | 67622 | No | NA | 143 | 25 | 0 | 高质量 | 高质量 | 100 | 基于 AAI(高置信度) | NA | 0 | 1.76 | 高 kmer_freq |
| 5 | 98051 | No | NA | 158 | 27 | 1 | 完整 | 高质量 | 100 | 基于 AAI(高置信度) | DTR | 0 | 1 |
在上述示例中,有 6 个病毒 contig 的结果
- 第一个 5325 bp 的 contig 没有完整性预测,这在 'checkv_quality' 字段中用 'Not-determined' 表示。这个 contig 也没有识别到病毒基因,所以它可能甚至不是病毒。
- 第二个 41803 bp 的 contig 被归类为 '低质量',因为其完整性 <50%。这个估计是基于 'AAI' 方法。请注意,仅在 quality_summary.tsv 文件中报告高或中等置信度的估计。您可以在 completeness.tsv 中查看更多详细信息。这个 contig 有一个 DTR,但被标记了某种原因(请参阅 complete_genomes.tsv 以获取详细信息)
- 第三个 contig 被认为是 '中等质量',因为其完整性估计为 80%,这是基于 'HMM' 方法。这意味着它太新颖,无法基于 AAI 估计完整性,但它与 CheckV 参考基因组共享 HMM。请注意,这个值代表下限(这意味着真实完整性可能高于这个值但不会低于这个值)。请注意,这个 contig 也被归类为前病毒。
- 第四个 contig 被归类为 '高质量',因为其完整性 >90%。然而,请注意 'kmer_freq' 的值为 1.7。这表明病毒基因组在 contig 中重复多次。这些情况相当罕见,但需要注意。
- 第五个 contig 被归类为 '完整',因为存在直接末端重复(DTR),根据 AAI 方法估计完整性为 100%。这个序列可以确信地被处理为一个完整基因组。
completeness.tsv
详细概述了完整性是如何估计的
| contig_id | contig_length | proviral_length | aai_expected_length | aai_completeness | aai_confidence | aai_error | aai_num_hits | aai_top_hit | aai_id | aai_af | hmm_completeness_lower | hmm_completeness_upper | hmm_hits |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 9837 | 5713 | 53242.8 | 10.7 | high | 3.7 | 10 | DTR_517157 | 78.5 | 34.6 | 5 | 15 | 4 |
| 2 | 39498 | NA | 37309 | 100.0 | medium | 7.7 | 11 | DTR_357456 | 45.18 | 30.46 | 75 | 100 | 22 |
| 3 | 29224 | NA | 44960.1 | 65.8 | low | 15.2 | 17 | DTR_091230 | 39.74 | 19.54 | 52 | 70 | 10 |
| 4 | 23404 | NA | NA | NA | NA | NA | 0 | NA | NA | NA | NA | NA | 0 |
在上述示例中,有 4 个病毒 contig 的结果
- 第一个前病毒 contig 使用基于 AAI 的方法估计的完整性为 10.7%(100 x 5713 / 53242.8)。这个估计的置信度很高,基于 3.7% 的相对估计误差,这反过来又基于 aai_id(平均氨基酸身份)和 aai_af(contig 对齐分数)与 CheckV 参考DTR_517517的相关性
- 第二个连续片段使用基于AAI的方法具有100%的完整性,使用基于HMM的方法具有75% - 100%的完整性范围。请注意,连续片段的长度略长于预期的基因组长度37,309碱基对。
- 第三个连续片段根据AAI方法估计为65.8%完整。然而,由于aai_confidence较低(意味着基于AAI的最顶部匹配相对较弱),我们不太信任这个结果。为了保守起见,我们可能希望根据HMM方法报告完整性范围(52-70%)。
- 最后一个连续片段没有基于AAI的匹配,也没有任何病毒HMM,因此我们无法对这个序列说任何话。
contamination.tsv
关于污染估计的详细概述
| contig_id | contig_length | total_genes | 病毒基因 | 宿主基因 | provirus | proviral_length | host_length | region_types | region_lengths | region_coords_bp | region_coords_genes | region_viral_genes | region_host_genes |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 98051 | 158 | 27 | 1 | No | NA | NA | NA | NA | NA | NA | NA | NA |
| 2 | 38254 | 54 | 23 | 2 | Yes | 36072 | 2182 | host,viral | 1-2182,2183-38254 | 1-2182,2183-38254 | 1-4,5-54 | 0,23 | 2,0 |
| 3 | 6930 | 9 | 1 | 2 | Yes | 3023 | 3907 | viral,host | 3023,3907 | 1-3023,3024-6930 | 1-5,6-9 | 1,0 | 0,2 |
| 4 | 101630 | 103 | 7 | 24 | Yes | 28170 | 73460 | host,viral,host | 46804,28170,26656 | 1-46804,46805-74974,74975-101630 | 1-43,44-85,86-103 | 0,7,0 | 13,0,11 |
在上述示例中,有 4 个病毒 contig 的结果
- 第一个连续片段不是一个预测的前病毒
- 第二个连续片段有一个覆盖2182碱基的预测宿主区域
- 第三个6930碱基对的连续片段在左侧有一个已识别的宿主区域
- 第四个101630碱基对的连续片段有103个基因,包括7个病毒和24个宿主基因。CheckV识别了两个宿主-病毒边界
complete_genomes.tsv
关于已识别的潜在完整基因组的详细概述
| contig_id | contig_length | prediction_type | confidence_level | confidence_reason | repeat_length | repeat_count |
|---|---|---|---|---|---|---|
| 1 | 44824 | DTR | high | 基于AAI的完整性 > 90% | 253 | 2 |
| 2 | 38147 | DTR | low | 低复杂度TR;重复TR | 20 | 10 |
| 3 | 67622 | DTR | low | 检测到多个基因组副本 | 26857 | 2 |
| 4 | 5477 | ITR | medium | 基于AAI的完整性 > 80% | 91 | 2 |
| 5 | 101630 | 前病毒 | 未确定 | NA | NA | NA |
在上面的例子中,有5个病毒连续片段的结果
- 第一个病毒连续片段有一个253碱基的直接终端重复序列。它被归类为高置信度,基于估计的完整性 > 90%
- 第二个病毒连续片段有一个20碱基的DTR,但由于DTR的低复杂度且不可信,导致置信度较低。DTR也发生了10次,被视为重复的。
- 第三个病毒连续片段有26857 bp的DTR!这表明基因组中有很大一部分是重复的。CheckV将其归类为低置信度,但用户可能需要手动解决这些重复
- 第四个病毒连续片段有91 bp的ITR。这被认为具有中等置信度,基于基于AAI的完整性 > 80%
- 第五个病毒连续片段在两侧都有宿主(前病毒)。然而,CheckV无法评估完整性,因此置信度保持为未确定
常见问题
问题:我可以用CheckV进行病毒预测吗?答:CheckV报告两种类型的病毒特征:(1)基因级针对病毒或宿主HMM的匹配,以及(2)基因组级与CheckV数据库中病毒的匹配。这两种类型的信息当然对于区分病毒和非病毒是有用的。然而,为了正确进行病毒预测,我们建议使用geNomad:https://github.com/apcamargo/genomad
问题:基于AAI和基于HMM的完整性有什么区别?答:基于AAI的完整性产生一个单一的估计完整性值,该值设计得非常准确,在报告的置信水平为中等或高时可以信赖。基于HMM的完整性给出完整性的90%置信区间(例如,30-75%),在这种情况下基于AAI的完整性不可靠。在这个例子中,我们可以有90%的把握(理论上)完整性在30%到75%之间。
问题:kmer_freq字段的意义是什么?答:这是病毒基因组在连续片段中呈现次数的度量。大多数情况下,这非常接近1.0。在罕见的情况下,组装错误可能导致连续片段序列代表病毒基因组的多个连续副本。在这些情况下,基因组_copies将超过1.0。
问:为什么我的DTR拼接体的估计完整性小于100%?答:如果估计的完整性接近100%(例如90-110%),则查询可能已完整。然而,有时不完整的基因组片段可能包含直接终端重复(DTR),在这种情况下,我们应预计它们的估计完整性小于90%,有时甚至更少。在其他情况下,拼接体可能确实是环形的,但估计的完整性是错误的。这也可能发生在查询到 multipartite 基因组的一个完整片段时(RNA 病毒很常见)。默认情况下,CheckV 使用90%的完整性截止值进行验证,但用户在这些模糊情况下可能希望自行判断。
问:为什么我的序列被认为是“高质量”的,尽管它有高污染?答:CheckV 仅根据完整性来确定序列质量。宿主污染很容易去除,因此不计入这些质量等级。
问:我进行了分箱并生成了病毒MAGs。我可以在这些上使用CheckV吗?答:CheckV 可以估计完整性但不能估计污染。在运行CheckV之前,您需要将每个MAG的拼接体连接成一个单一序列。
问:我可以用CheckV从全基因组(元基因组)预测(前)病毒吗?答:可能可以,尽管这还没有经过测试。相反,我们建议使用geNomad: https://github.com/apcamargo/genomad
问:如何处理没有完整性估计的假“封闭基因组”?答:在某些情况下,您可能无法验证具有终端重复或前病毒整合位点的序列的完整性。DTR是相当可靠的指标(>90%的时间),在没有完整性估计的情况下可能可以信赖。然而,完整的前病毒和ITR是可靠性较低的指标,因此需要>90%的估计完整性。
问:为什么我的拼接体被分类为“质量未确定”?答:这发生在序列与CheckV参考基因组没有足够相似度以自信地估计完整性,并且没有病毒HMM的情况下。这种情况可能有几种解释,按可能性顺序排列:1) 您的拼接体非常短,并且偶然情况下它没有与CheckV参考共享任何基因,2) 您的拼接体来自一个非常新颖的病毒,它与CheckV数据库中的所有基因组都亲缘关系较远,3) 您的拼接体根本不是病毒,因此与任何参考都不匹配。
问:如何处理“质量未确定”的序列?答:虽然无法估计这些序列的完整性,但您可以选择分析长度超过一定值(例如>30 kb)的序列。
问:为什么包括0个病毒基因的序列出现在CheckV输出中?答:目前,CheckV假设输入序列代表病毒,并试图估计其质量。一些输入序列可能来自细菌、质粒或其他来源,因此可能检测到0个病毒基因。
支持代码
基于成对ANI的快速基因组聚类
首先,创建一个blast+数据库
makeblastdb -in <my_seqs.fna> -dbtype nucl -out <my_db>
接下来,使用blast+包中的megablast进行所有序列的全对全blastn
blastn -query <my_seqs.fna> -db <my_db> -outfmt '6 std qlen slen' -max_target_seqs 10000 -o <my_blast.tsv> -num_threads 32
接下来,通过组合序列对的局部比对来计算成对ANI
anicalc.py -i <my_blast.tsv> -o <my_ani.tsv>
最后,使用MIUVIG推荐参数(95% ANI + 85% AF)执行UCLUST-like聚类
aniclust.py --fna <my_seqs.fna> --ani <my_ani.tsv> --out <my_clusters.tsv> --min_ani 95 --min_tcov 85 --min_qcov 0
文件 <my_clusters.tsv> 包含聚类结果。第一列是集群代表,第二列包含集群成员。文件 <my_ani.tsv> 包含成对ANI结果。列包括
- query_id:查询标识符
- target_id:checkv参考基因组标识符
- alignment_count:blastn比对数量
- ani:平均核苷酸身份
- query_coverage:比对覆盖查询基因组的百分比
- target_coverage: 目标基因组由比对覆盖的百分比
引用
如果您在研究中使用了该软件,请引用
Nayfach, S., Camargo, A.P., Schulz, F. et al. CheckV评估了基于宏基因组组装的病毒基因组的质量和完整性。自然生物技术 39, 578–585 (2021)。https://doi.org/10.1038/s41587-020-00774-7


下载文件
下载适用于您平台的文件。如果您不确定选择哪个,请了解有关安装包的更多信息。