基因组区间数据帧的操作和工具。



项目描述
Bioframe:对基因组区间数据帧的操作








Bioframe允许在Python中对基因组区间数据帧进行灵活和可扩展的操作。
Bioframe直接建立在Pandas之上。Bioframe提供
- 各种在数据帧上直接工作的基因组区间操作。
- 针对基因组区间的特殊类别进行操作,包括染色体臂和固定大小的区间。
- 针对多样化的表格基因组数据格式和加载基因组组装摘要信息的功能。
Bioframe是NumFOCUS的附属项目。
安装
pip install bioframe
贡献
有兴趣为bioframe做出贡献?太好了!要开始,请查看贡献指南。关于项目路线图讨论在Open2C Slack上进行,并在此安排了常规的开发者会议。任何人都可以加入并参与!
区间操作
bioframe中的关键基因组区间操作包括
overlap:在两个数据框之间找到重叠的基因组区间对。closest:对于数据框中的每个区间,找到第二个数据框中最近的区间。cluster:将数据框中的重叠区间分组到簇中。complement:找到任何数据框区间都不覆盖的基因组区间。
Bioframe还包含用于基因组区间操作并经常使用的函数,可以表示为核心操作和数据框操作的组合,包括:coverage、expand、merge、select和subtract。
要调用overlap两个数据框,请
import bioframe as bf
bf.overlap(df1, df2)
对于这两个输入数据框,所有区间都在同一染色体上
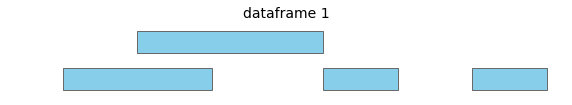

overlap将返回以下区间对作为重叠
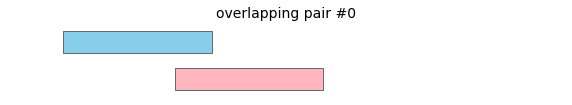
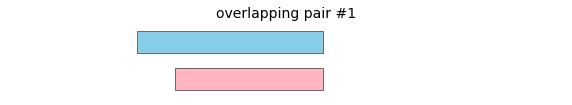
要合并数据框中所有重叠的区间,请调用
import bioframe as bf
bf.merge(df1)
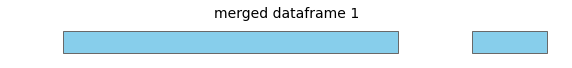
对于这个输入数据框,所有区间都在同一染色体上
merge将返回一个新的数据框,其中包含这些合并的区间
查看指南以了解bioframe中其他区间操作的可视化。
文件输入/输出
Bioframe包括将基因组文件格式读入数据框以及反之亦然的实用程序。一个方便的函数是read_table,它类似于pandas的read_csv/read_table,但提供了一个schema参数,用于填充常见表格文件格式的列名。
jaspar_url = 'http://expdata.cmmt.ubc.ca/JASPAR/downloads/UCSC_tracks/2022/hg38/MA0139.1.tsv.gz'
ctcf_motif_calls = bioframe.read_table(jaspar_url, schema='jaspar', skiprows=1)
教程
请参阅这个jupyter笔记本,了解如何使用bioframe将TF基序分配给ChIP-seq峰。
引用
如果您在您的作品中使用了bioframe,请引用
@article{bioframe_2024,
author = {Open2C and Abdennur, Nezar and Fudenberg, Geoffrey and Flyamer, Ilya M and Galitsyna, Aleksandra A and Goloborodko, Anton and Imakaev, Maxim and Venev, Sergey},
doi = {10.1093/bioinformatics/btae088},
journal = {Bioinformatics},
title = {{Bioframe: Operations on Genomic Intervals in Pandas Dataframes}},
year = {2024}
}


下载文件
下载您平台对应的文件。如果您不确定该选择哪个,请了解更多关于 安装包 的信息。
源代码分布
bioframe-0.7.2.tar.gz (963.5 kB 查看哈希值)
构建分布
bioframe-0.7.2-py2.py3-none-any.whl (151.5 kB 查看哈希值)
关闭
bioframe-0.7.2.tar.gz 的哈希值
| 算法 | 哈希摘要 | |
|---|---|---|
| SHA256 | 23fa150948fb1f9409a8d608c94f222fd2e144c8f1ac965879517d5e87d2c598 |
|
| MD5 | 866af56db5fa6dda270e1cd10e28df19 |
|
| BLAKE2b-256 | a062dba524f00b36af9f2df52b65431510dd15af5b74f858ee5ca931da922a90 |
关闭
bioframe-0.7.2-py2.py3-none-any.whl 的哈希值
| 算法 | 哈希摘要 | |
|---|---|---|
| SHA256 | ee5aa0ee00cdd997aa304d7527b42563d6a0af5fd7eedf22da2224e6848dc3c8 |
|
| MD5 | 966322633a5d557f273d75211b9a3447 |
|
| BLAKE2b-256 | 313b6f58a614f3bbceafb69ec5a4126620abad253718ae21d66412c14f0c8b64 |